药物代谢动力学
作者:admin发布时间:2010-04-24 11:03浏览:
次

图2-1 机体对药物的处置
一、药物分子的跨膜转运
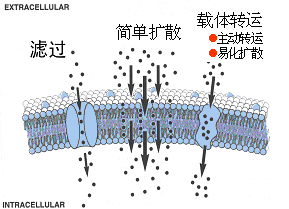
图2-2 药物通过细胞膜的方式
(一)单纯扩散 (simple diffusion,passive diffusion)
1. 概念:脂溶性物质直接溶于膜的类脂相而通过
2. 特点: *转运速度与药物脂溶度(lipid solubility)成正比
*顺浓度差,不耗能。
*转运速度与浓度差成正比
*转运速度与药物解离度 (pKa) 有关
*离子障(ion trapping):
分子 极性低,疏水,溶于脂,可通过膜
离子 极性高,亲水,不溶于脂,不通过
pH和pKa决定药物分子解离多少
酸性药:Ka =[ H+ ] [ A- ]/[HA] pKa = pH - log 10pH-pKa =
碱性药:Ka =[ H+ ] [ A- ]/[HA] pKa = pH - log 10pKa- pH =
(二)滤过(filtration)
1. 概念:水溶性小分子药物通过细胞膜的水通道,受流体静压或渗透压的影响
(三)主动转运 (active transport)
1. 概念:需依赖细胞膜内特异性载体转运,如 5-氟脲嘧啶、甲基多巴等
2. 特点:*逆浓度梯度,耗能
*特异性(选择性)
*饱和性
*竞争性
(四)易化扩散(facilitated diffusion )
1. 概念:在膜蛋白的帮助下物质从高浓度侧向低浓度侧跨膜转运
2. 特点:*需特异性载体
*顺浓度梯度,不耗能
二、药物的体内过程
(一)吸收 (Absorption)
1. 概念:从给药部位进入全身循环
2. 分类:
(1) 口服给药 (Oral ingestion)
*吸收部位主要在小肠
*停留时间长,经绒毛吸收面积大
*毛细血管壁孔道大,血流丰富
*PH5-8,对药物解离影响小
Fick扩散律 (Fick’s Law of Diffusion)
流量 (单位时间分子数) = 面积 ′ 通透系数/厚度
首过消除 (first pass eliminaiton)
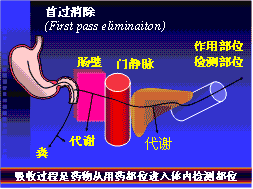
图2-3 首过消除示意图
(2) 静脉注射给药(intravenous)
(3) 肌肉注射和皮下注射 (intramuscular and subcutaneous injection)
*被动扩散+过滤,吸收快而全
*毛细血管壁孔半径40?,大多水溶性药可滤过
(4) 呼吸道吸入给药 (inhalation)
*气体和挥发性药物(全麻药)直接进入肺泡,吸收迅速
*肺泡表面积大(100-200m2)
*血流量大(肺毛细血管面积80 m2 )
(5) 经皮给药 (transdermal)
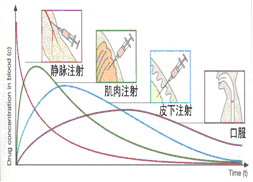
图2-4 给药方式与血药浓度的关系
(二)分布 (distribution)
1. 概念:药物从血循到达作用、储存、代谢、排泄等部位
2. 影响因素:
*脂溶度
*局部 pH 和药物离解度
*毛细血管通透性
*组织通透性
*转运蛋白量
*血流量和组织大小
*血浆蛋白和组织结合
血浆蛋白结合(plasma protein binding)
D+P DP
[DP]/ [PT]= [D]/( KD+[D])
影响因素:*可逆性(reversible equilibrium),结合量与D、PT和KD有关
*可饱和性(saturable)
*DP不能通过细胞膜
*非特异性和竞争性 (nonspecific & competitive)
血脑屏障 (blood-brain barrier, BBB)
特点:*大分子、脂溶度低、DP不能通过
*有中枢作用的药物脂溶度一定高
*也有载体转运,如葡萄糖可通过
*可变:炎症时,通透性↑,大剂量青霉素有效
胎盘屏障 (placental barrier)
特点:*胎毛细血管内皮对药物转运的选择性-
*脂溶度、分子大小是主要影响因素 (
*母血pH = 7.44; 胎血pH=7.30。弱碱性药物在胎血内易离解
*胎盘有代谢(如氧化)药物的功能
*转运方式和其它细胞相同:简单扩散
*大多数药物均能进入胎儿
(三)代谢(生物转化, metabolism, biotransformation):
步骤:分两步反应
I期反应(Phase I):氧化、还原、水解引入或脱去基团(-OH、-CH3、-NH2、-SH)
II期反应(Phase II):内源性葡萄糖苷酸、硫酸、醋酸等与药物或I期反应的代谢物结合生成极性很高的代谢产物
药物氧化代谢 (oxidation):主要是由细胞色素P450单氧化酶系代谢

图2-5 细胞色素P450单氧化酶系示意图
(四)排泄 (excretion):
途径:肾脏(主要),消化道,肺,皮肤,唾液,乳汁等
三、房室模型
1. 概念:视身体为一系统,按动力学特点分若干房室;为假设空间,与解剖部位或生理功能无关;转运速率相同的部位均视为同一房室;因药物可进、出房室,故称开放性房室系统;开放性一室模型和开放性二室模型为常见。
2.分类:
一房室模型:体内药物瞬时在各部位达到平衡,即给药后血液中依度和全身各组织器官部位浓度迅即达到平衡
二房室模型:药物在某些部位的药物浓度和血液中的浓度迅速达平衡,而在另一些部位中的转运有一速率过程,但彼此近似,前者被归并为中央室,后者则归并成为外周室
三房室模型:转运到外周室的速率过程有较明显快慢之分
3.影响房室模型判断的因素:
*用药途径:如茶碱口服为一室;静注为二室
*药物吸收速率:如药物分布迅速,则口服后吸收过程中即包含分布过程,无分布相可见
*采血总时程:间隔时间不当可遗漏房室
*采血点的数量和分布
*分析方法的敏感性
二室模型计算公式:C=Ae-a t+Beb t
C: t 时血浆药物浓度;a: 分布速率常数;b: 消除速率常数;B b相外延至纵轴的截距;A 实测浓度和b相各相应 t 时浓度之差形成的直线在纵轴上的截距;e: 自然对数之底=2.718
四、药物消除动力学
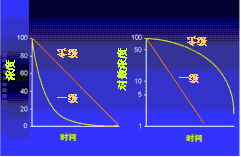
图2-6 体内药物浓度因不断消除而随时间不断变化,dC/dt = - kCn
分类:一级消除动力学 (first order elimination kinetics ): n = 1 dC/dt = - kC
转运(消除)速度与浓度差成正比
零级消除动力学 (zero order elimination kinetics): n = 0 dC/dt = k
药物达一定浓度,机体消除能力达最大后的消除动力学;因消除能力饱和,单位时间消除药量不变,消除速度不再与药物浓度有关
五、体内药物的药量-时间关系
1. 概念:
① 一次给药: 峰浓度(Cmax):一次给药后的最高浓度,此时吸收和消除达平衡。
达峰时间(Tmax):给药后达峰浓度的时间,多为2(1-3)hrs。
曲线下面积(AUC): 药-时曲线下所覆盖的面积,其单位为:ng×h/mL ,反映药物体内总量。
② 多次给药: 稳态血药浓度 (steady-state concentration):按照一级动力学规律消除的药物,其体内药物总量随着不断给药而逐步增多,直至从体内消除的药物量和进入体内的药物量相等时,体内药物总量不再增加而达到稳定状态,此时的血浆药物浓度称为稳态血药浓度。
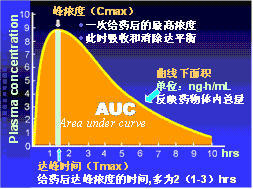

图2-7、8 药-时曲线相关概念示意图
六、药物代谢动力学重要参数
1.消除半衰期(half-life, T1/2):血浆药物浓度消除一半所需时间
一级消除动力学 T1/2 = 0.693/k,T1/2与浓度无关,为恒定值。
零级消除动力学 T1/2 = 0.5 ′ C0/k
2.清除率 (clearance):单位时间内多少容积血浆中的药物被清除,反映肝肾功能.单位:L/h或ml/min CL=CL肾脏+CL肝脏+CL其它;计算公式: CL = D/AUC
3.表观分布容积 (volume of distribution):体内药物总量和血浆药物浓度之比,Vd=D/C,Vd非体内生理空间.其意义是:推测药物在体内的分布范围,计算用药剂量:Vd=D/C。
4.生物利用度(Bioavailability):药物到达全身血循环内的相对量和速度
绝对生物利用度:F = (AUC血管外/AUC血管内) × 100%
相对生物利用度:F = (AUC受试制剂 ¤ AUC标准制剂) × 100%
七、药物剂量的设计和优化
1.靶浓度(target concentration)
2.维持量(maintenance dose)
3.负荷量(loading dose):首次剂量加大,然后再给予维持剂量,使稳态治疗浓度提前产生。
4.个体化治疗(individualized treatment)